![]()




























![]()
![]()
研究成果 Research Results
- TOP
- News
- Research Results
- When less is more: A promising approach for low-cell-number epigenomic profiling
When less is more: A promising approach for low-cell-number epigenomic profiling
2018.12.10Research ResultsLife & Health
Scientists at Kyushu University and Tokyo Institute of Technology in Japan have developed a technique that enables analysis of DNA–protein interactions using very small numbers of cells, ranging from 100 to 1,000. Their method could capture previously unexamined epigenomic information, facilitate biomarker discovery and open new avenues for precision medicine.
A collaborative study by researchers at Kyushu University, Tokyo Institute of Technology, Waseda University, the University of Tokyo and Osaka University has led to the development of a unique approach to epigenomic profiling that involves working with much fewer cells than in existing methods.
The technique, called Chromatin Integration Labeling sequencing (ChIL-seq), could open up new opportunities for scientists to study rare cell types and other cell samples in short supply. ChIL-seq requires only a fraction of starting cellular material. In a series of experiments to evaluate ChIL-seq performance, the researchers successfully demonstrated detection of histone modifications and DNA-binding factors using just 100 to 1,000 cells.
For the past decade, chromatin immunoprecipitation sequencing (ChIP-seq) has been the dominant technique for analyzing epigenomic data and identifying important binding sites of DNA-associated proteins. However, one limitation has been that ChIP-seq requires at least 10,000 or typically millions of cells to start off with, mainly due to the fact that samples tend to be lost during two key steps: chromatin preparation and immunoprecipitation.
The research team, co-led by Hiroshi Kimura of the Institute of Innovative Research, Tokyo Institute of Technology, and Yasuyuki Ohkawa of Medical Institute of Bioregulation, Kyushu University, overcame the problem of sample loss by replacing the two steps mentioned above with immunostaining, a non-destructive method suitable for analyzing tissue specimens.
Using ChIL-seq, the team also detected genomic regions associated with histone marks at the single-cell level—an achievement that brings biologists closer to the long-held goal of establishing single-cell profiling.
ChIL-seq can “zoom in” on genomic sequences near target molecules before cellular breakdown, and this is particularly useful for studying adherent cells, i.e., whole cells that remain attached to culture plates and after immunofluorescence.
Many different types of epigenome profiling methods are now being developed around the world. Each has its advantages and limitations. The researchers point out that ChIL-seq, too, still needs refining. In its current form, for example, it has low sensitivity to heterochromatin regions, and it can take 3–4 days to complete the procedure.
Overall, they are confident that ChIL-seq holds promise due to its precision, which makes it suitable for single-cell applications, and its flexibility, meaning that in future, it could be combined with other powerful sequencing techniques.
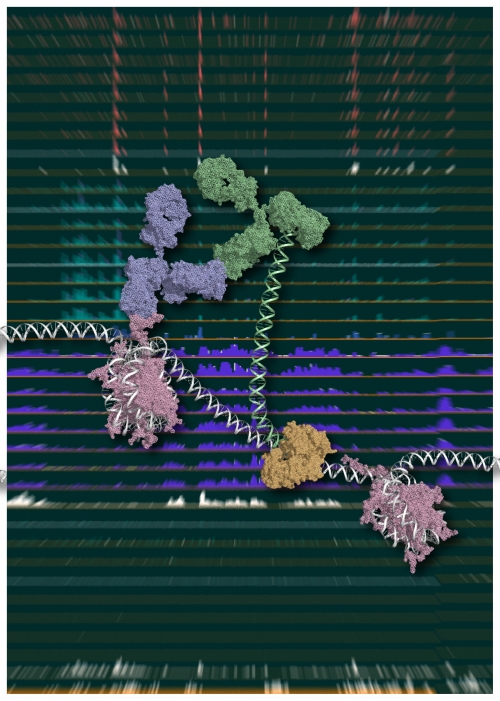
Figure 1.
Chromatin Integration labeling (ChIL)
Schematic model of ChIL reaction.
The oligonucleotide conjugated with the sec-ond-ary antibody (green), which binds to the primary antibody (blue) recognizing a specific medication of histone (purple), is integrated into proximal genomic DNA by Tn5 transposase (brown).
An image courtesy of Yuko Sato (Tokyo Institute of Technology).
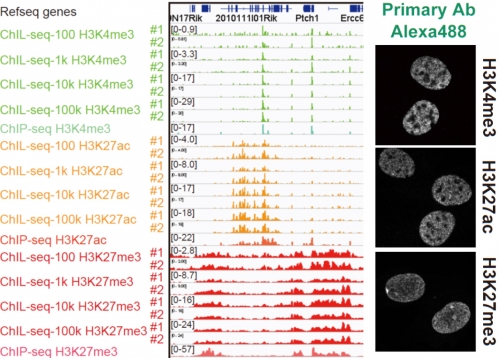
Figure 2.
Chromatin Integration labeling sequencing
Histone modifications or transcription factors are labeled with ChIL probe in cell nuclei (right), and the genomic sequence proximal to the probe is amplified for large scale sequencing analysis (left). The profiles observed in 100 cells are similar to those using more than 1,000 cells.
Researcher comments
We as epigenome researchers had been in great need of this technology. Though we established a new conception earlier, it took us more than five years to eventually become a practical technique. We strongly hope this technology will help great leap in life science, including the regenerative medicine by stem cells and elucidation of mechanism of cancers.
Journal Reference
A chromatin integration labelling method enables epigenomic profiling with lower input, ,Nature Cell Biology , 10.1038/s41556-018-0248-3Research-related inquiries
- TOP
- News
- Research Results
- When less is more: A promising approach for low-cell-number epigenomic profiling