![]()




























![]()
![]()
研究成果 Research Results
- TOP
- News
- Research Results
- An innovative technology to simultaneously detect multiple epigenetic information in single specimens
An innovative technology to simultaneously detect multiple epigenetic information in single specimens
2020.09.14Research ResultsLife & Health
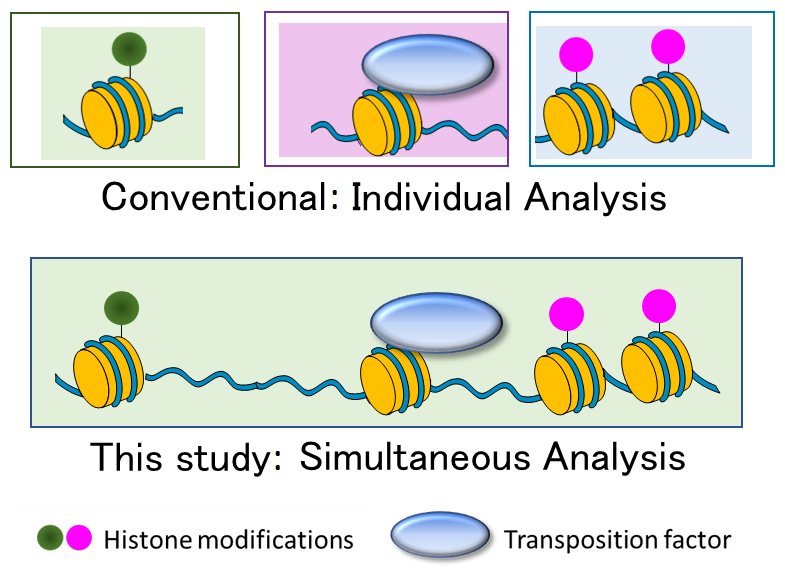
Conventionally, multiple epigenomes have been analyzed individually (top), but the mtChIL method enables analysis of multiple epigenomes at the same time.
Researchers from Japan have now reported the detailed protocol for a low-input process to analyze the epigenome—the set of molecules interacting with DNA to select which genes are expressed or repressed and thereby determine cell functionality. The group also developed a new application of the technology to simultaneously detect multiple epigenomic information in single specimens, which will help spur advances in areas related to cancer research and regenerative medicine.
Composed of 30 trillion cells, the human body is an extremely complex machine with nearly everyone of its cells depending on the same genome to accomplish all of the body’s incredible feats. However, while the genome coded in the DNA may be the same for different cells, interactions of molecules with DNA to select which genes from the tens of thousands of the genome to express without modifying the underlying code are essential for achieving the cell-type specific functions of the over 200 types of cells in the body.
Called the epigenome, these chemical compounds help choose which genes to express or repress, so deciphering epigenomic information and its regulatory mechanisms is critical for understanding how genes are selected for their expression during development and differentiation and for making advances in areas such as tissue regeneration and stem cell medicine. While obtaining epigenomic information from single cells is ideal, low-input epigenome profiling requires a cutting-edge technology with high skills.
Researchers from Kyushu University, Tokyo Institute of Technology, and the Institute for Quantitative Biosciences at the University of Tokyo are now hoping to accelerate the broad adoption of low-input analysis methods through the publishing of a detailed step-by-step protocol using the Chromatin Integration Labeling (ChIL) method, one of the most sensitive technologies. Furthermore, the group has applied the method to develop Multitarget ChIL, or mtChIL, which can simultaneously acquire multiple epigenetic information, e.g., a histone modification and a transcription factor, from the same samples.
This technology enables comprehensive analysis of combinations of factors that could previously only be analyzed individually, making it possible to develop more precise epigenetic genome editing with applications in various fields including cancer—which is a disruption in gene expression—and regenerative medicine—which requires specific induction of gene expression.
###
For more information about this research, see “Chromatin integration labeling for mapping DNA-binding proteins and modifications with low input,” Tetsuya Handa, Akihito Harada, Kazumitsu Maehara, Shoko Sato, Masaru Nakao, Naoki Goto, Hitoshi Kurumizaka, Yasuyuki Ohkawa, and Hiroshi Kimura, Nature Protocols (2020). https://doi.org/10.1038/s41596-020-0375-8
The research was supported in part by the "Japan Science and Technology Agency (JST), Strategic Research for Evolutional Science and Technology (PI: Yasuyuki Ohkawa)," "JST PRESTO Genome programming (PI: Akihito Harada)," and the "Chromatin Potential" project of the Ministry of Education, Culture, Sports, Science, and Technology (PI: Hiroshi Kimura).
Research-related inquiries
Yasuyuki Ohkawa, Professor
Division of Transcriptomics, Medical Institute of Bioregulation
Contact information can also be found in the full release.
- TOP
- News
- Research Results
- An innovative technology to simultaneously detect multiple epigenetic information in single specimens