![]()




























![]()
![]()
研究成果 Research Results
- TOP
- News
- Research Results
- Uncovering the genetic mechanism behind Rett syndrome
Uncovering the genetic mechanism behind Rett syndrome
Dysfunction in key gene causes neural stem cells to produce more astrocytes than neurons 2021.07.02Research ResultsLife & Health
Medical researchers led by Kyushu University have revealed a possible underlying genetic pathway behind the neurological dysfunction of Rett syndrome. The team found that deficiencies in key genes involved in the pathology triggers neural stem cells to generate less neurons by producing more astrocytes—the brain's maintenance cells.
The researchers hope that the molecular pathology they identified, as reported in the journal Cell Reports, can lead to potential therapeutic targets for Rett syndrome in the future.
Rett syndrome is a progressive neurodevelopmental disorder characterized by impairments in cognition and coordination—with varying severity—and occurs in roughly one in every 10,000 to 15,000 female births. However, it is difficult to initially identify because children appear to develop normally in the first 6–18 months.
"Rett syndrome is caused by mutations in a single gene called methyl-CpG binding protein 2, or MeCP2. The gene was identified over two decades ago and much has been uncovered since, but exactly how the mutations cause the pathology remains elusive," explains first author Hideyuki Nakashima of Kyushu University’s Faculty of Medical Sciences.
In their past research, the team had identified that MeCP2 acts as a regulator for the processing of specific microRNAs to control the functions of neurons. So, they went back to investigate if that pathway was also involved in the differentiation of neural stem cells.
Compared to messenger RNA, the final template transcribed from DNA that is used by a cell to synthesize proteins, microRNAs—or miRNAs—are much smaller and act to regulate messenger RNA to make sure the cell is making the correct amount of the desired protein.
"Through our investigation, we found several microRNAs associated with MeCP2, but only one affected the differentiation of neural stem cells: a microRNA called miR-199a," says Nakashima. "In fact, when either MeCP2 or miR-199a are disrupted, we found that it increased the production of cells called astrocytes."
Astrocytes are like the support cells of your brain. While neurons fire off the electrical signals, astrocytes are there to help maintain everything else. During development, astrocytes and neurons are generated from the same type of stem cells, known as neural stem cells, where their production is carefully controlled. However, dysfunction in MeCP2 or miR-199a causes these stem cells to produce more astrocytes than neurons.
"Further analysis showed that miR-199a targets the protein Smad1, a transcription factor critical for proper cellular development. Smad1 functions downstream of a pathway called BMP signaling, which is known to inhibit the production of neurons and facilitate the generation of astrocytes," states Nakashima.
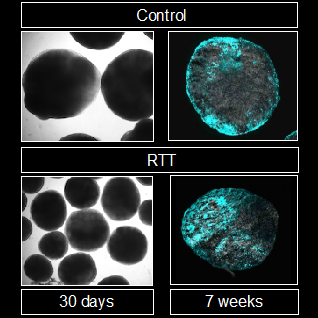
To investigate the process further, the team established a brain organoid culture—a 3D culture of neural stem cells that can mimic aspects of brain development—from iPS cells derived from patients with Rett syndrome. When they inhibited BMP, short for bone morphogenetic protein, the team was able to reduce abnormal neural stem cell differentiation.
"Our findings have given us valuable insight into the role of MeCP2, miR-199a, and BMP signaling in the pathology of Rett syndrome," concludes Kinichi Nakashima, who headed the team. "Further investigation is needed, but we hope this can lead to clinical treatments for Rett syndrome symptoms."
###
For more information about this research, see "MeCP2 controls neural stem cell fate specification through miR-199a-mediated inhibition of BMP-Smad signaling," Hideyuki Nakashima, Keita Tsujimura, Koichiro Irie, Takuya Imamura, Cleber A. Trujillo, Masataka Ishizu, Masahiro Uesaka, Miao Pan, Hirofumi Noguchi, Kanako Okada, Kei Aoyagi, Tomoko Andoh-Noda, Hideyuki Okano, Alysson R. Muotri, and Kinichi Nakashima, Cell Reports (2021). https://doi.org/10.1016/j.celrep.2021.109124
This release is also available in Japanese.
Research-related inquiries
Kinichi Nakashima, Professor
Department of Stem Cell Biology and Medicine, Faculty of Medical Sciences
Contact information can also be found in the full release.
- TOP
- News
- Research Results
- Uncovering the genetic mechanism behind Rett syndrome